Table of contents
- Multiple sequence alignments
- Specificity determinant residues
- CONAN
- Results reports
- Example
- Browser Compatibility
Multiple Sequence Alignments
Multiple Sequence Alignments (MSA) consist of a set of at least three biological sequences (proteins, RNA or DNA) aligned in a table format so that respective positions of the same sequence assume the same column in the table. If the sequences have a homological relationship, it is possible to say that the patterns of amino acid variability in each column represent a manifestation of substitutions under constraints imposed by the function [Dima & Thirumalai, 2006]. However, a MSA tells an evolutionary history according to a series of events such as evolutionary pressure, mutations, recombination and genetic drift [Valdar, 2002].
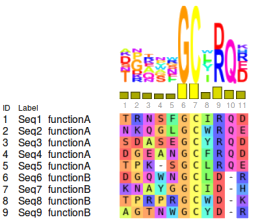
As expected by the Neutral Theory of Molecular Evolution [Kimura, 1968], most columns of a MSA usually exhibit high amino acid variability, probably related to neutral substitutions. In contrast, a few columns present amino acids that were strictly conserved during evolution, exhibiting an extremely low variability. The use of conserved positions as predictors of functional/structural importance predates the use of Multiple Sequence Alignments, and was proposed still in the 1960s by Zuckerkandl & Pauling [1965]. Conservation does not always occur in relation to a specific the amino acid, but also in relation to physical-chemical and structural properties that needs to be maintained so that the protein retains its activity and stability. This type of pattern is called marginally conserved positions [Chakrabarti et al., 2007].
Some biological processes, as the gene duplication followed by divergence, allows the emergence of proteins with activity completely different from their ancestors. This is due to the fact that after duplication, one of the copies may lose its evolutionary constraints, since the production of that protein will be offset by the copy of the gene. Thus, previously prohibitive mutations begin to occur without any damage to the organism, and may lead to a process of neofunctionalization. Therefore, protein families can contain multiple functional subclasses, and the identification of locally conserved positions can also be related to specificity determinant residues.
Specificity Determinant Residues
Specificity Determinant Sites (SDS) consist of group of residues sufficiently capable of distinguish a subset of homologous sequences according to some kind of functional or structural shared feature. These patterns can be extracted from a multiple sequence alignments by observing sites that are higly conserved within a subclass but not in the whole alignment. Chakraborty & Chakrabarti [2014] introduced three types of Specificity Determinant Sites. Type 1 is related to functional divergence, when the subfamilies have distinct evolutionary constraints, the SDS of each class can be composed by amino acids in different positions. Type 2 is more related to details specificity, as an affinity to a specific ligand. It occurs when the position is conserved in multiple subclass, but the amino acid that define the specificity vary between them. Finally, SDS type 3 is related to marginally conserved residues, that is, physical-chemical or structural property constraints.
The detection of SDSs is usually performed with a small set of curated sequences, thus, one can split the predefined subfamilies and search for patterns that are specific of each of them. Given that the number of available curated sequences consists of a small fraction of all sequences deposited in the databases, and these sequences tend to be biased by economic and pharmacological interests, often there is insufficient sampling to accomplish this type of analyze. Therefore, we developed a method with the in order to use all this range of available sequences to detect possible SDS from the co-variation tendency of the residues [Fonseca, N et al., 2018].
CONAN
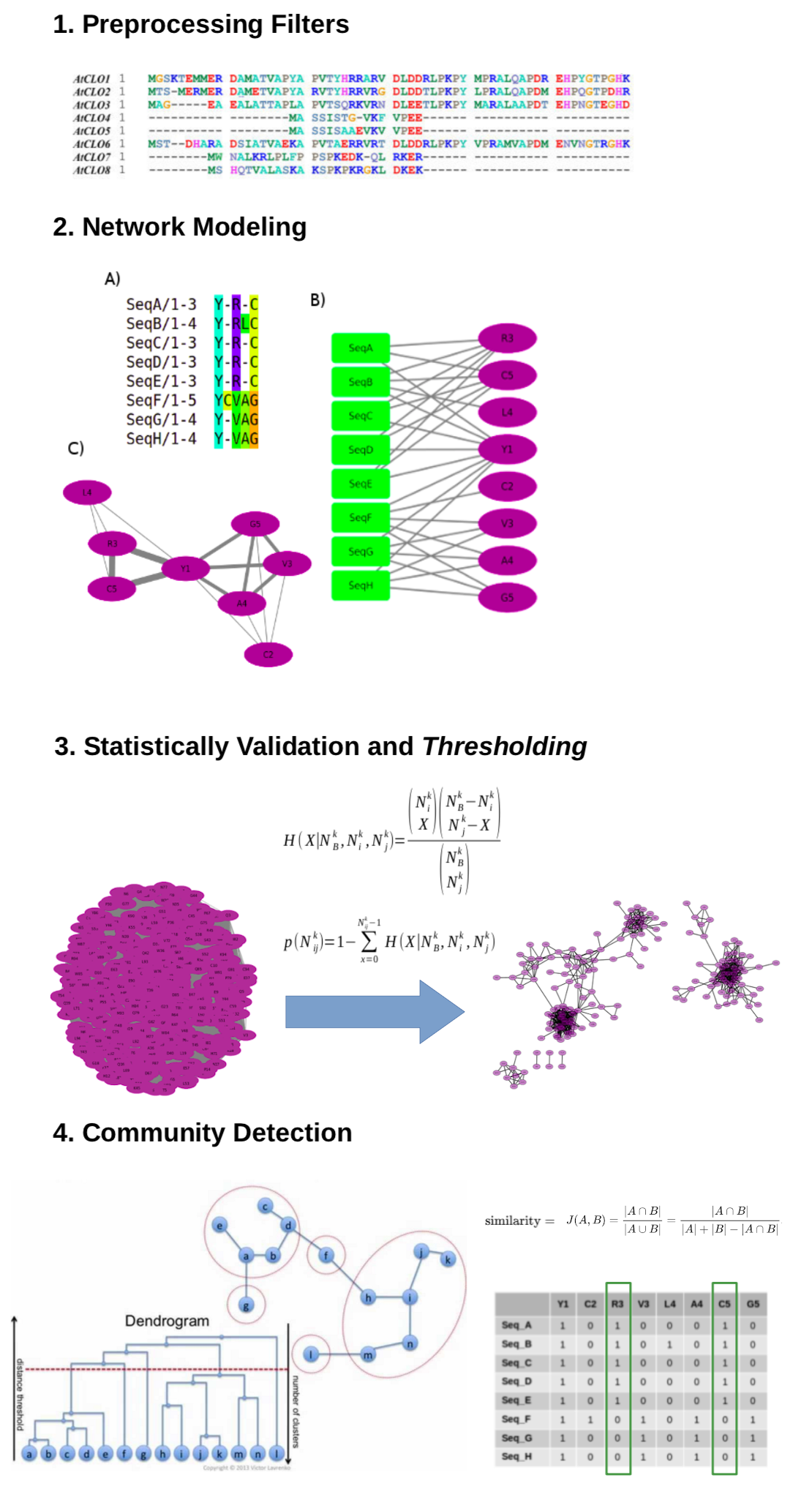
This software calculates and analyses a residue co-variation network with the aim to emphasize local co-evolutionary constraints and consequently detect specificity determinants and function related sites. Since this methodology works with large MSAs including unannotated sequences, a crucial step is pre-processing, in which fragments and sequences with high identity are removed. It is also possible remove from the analysis residues with very low frequencies, which do not have enough sampling to pass the randomness test and therefore can be discarded, saving computational time. Similarly, extremely conserved residues can also be discarded as such pattern detection is straightforward.
In this method, the MSA is represented by a bipartite graph, a network whose nodes can be divided in two disjoint and independent sets U and V. In the Multiple Sequence Network, U is composed by the sequence labels and V by all residues present in the alignment (i.e. an amino acid or stereochemical property followed by its position). By generating the monopartite projection of V, groups of co-occurring residues in the alignment will tend to form communities. This monopartite network is then submitted to statistical validation of the edges, redundancy filters, thresholding and finally community detection. Further details about the method can be obtained in a previous paper by Fonseca, N et al., 2019. The application can also perform the edge validation through the DRCN method [Bleicher, L. et al., 2011].
CONAN accepts four types of input: a multiple sequence alignment file in the formats selex, Stockholm or fasta; a single fasta sequence; a single UniProtKb ID or a Pfam ID. If the user submits a single sequence or an UniProtKb ID, a Hmmerscan will be performed and the user will be able to select which protein family is going to be analysed. All multiple sequence alignments are fetched from Pfam using the Full dataset.
This application applies statistical based analysis, thus the greater the number of sequences (after the filtering procedures), the better the confidence of the results obtained. Unfortunately, this web server currently has a restriction of input size: 50mb file and 4.000 sequences using the marginal conservation detection or 15.000 sequences otherwise, therefore, some inputs may not be suitable to be executed through the web application. For these cases, you can download our standalone application and run in your own computer or HPC cluster. In our tests, we were able to obtain accurate results from alignments contained at least 200 to 300 sequences after the filtering procedures.
CONAN Results
The CONAN results pages are session-based, so it loads all the required data on the first access, making it easy and fast to navigate between the several results topics. You may want to keep the URL of your analysis, since it allows you to return and share that information. If the page becomes inactive for a few minutes, the session will be interrupted, so you will need to access it again through your results address (e.g. http://www.biocomp.icb.ufmg.br:8080/conan/results?id=[your job id]).
The detected communities indicate sets of highly co-occurring residues in the alignment after the removal of fragments and high identity sequences. These residues are usually related to the specificity factors from subsets of sequences in the alignment. The CONAN results reports have a couple of features to assist you in the identification of the biological meaning of the detected patterns.
Home page
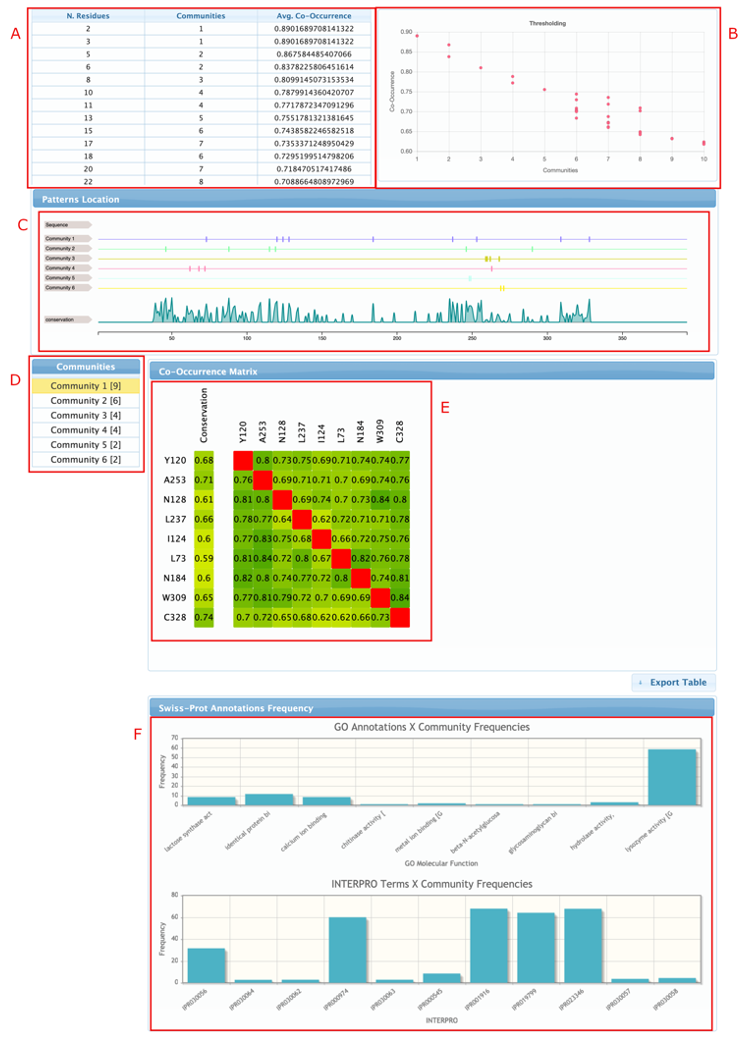
The home page of the results report is where the user can manually refine the parameters of the analysis and see how it affects the results. As can be seen in the figure, it has six main objects:
- The threshold controller, where the user can select new cut-off values for the edges making the network more stringent or flexible.
- The distribution of number of detected communities by the average co-variation score.
- Patterns location shows where the detected residues are located in the sequences and how conserved they are. It also shows the consensus sequence (usually hidden by default, but can be shown after zooming in – right click to reset the zoom)
- Communities menu is used to navigate through the detected communities. Changing the selected communities will update the co-occurrence matrix and the annotations charts.
- Co-occurrence matrix. The first column (conservation) shows the overall frequency of each residue of the selected community in the filtered multiple sequence alignment, the rest of the fields shows the frequency of residue X given that residue Y is present in the alignment (e.g. Y120 is present in 68% of the sequences of the filtered alignment, but in 80% of the sequences that also have A253; in the same way, A253 occurs in 71% of the sequences of the filtered alignment and in 76% of the sequences that also have Y120.
- Annotations frequency charts shows the percentage of residues from the selected community that are presented in sequences from the UniProtKb/Swiss-Prot that have each of the Gene Ontology and INTERPRO terms. This is useful to identify communities that are specific to some functional subclass.
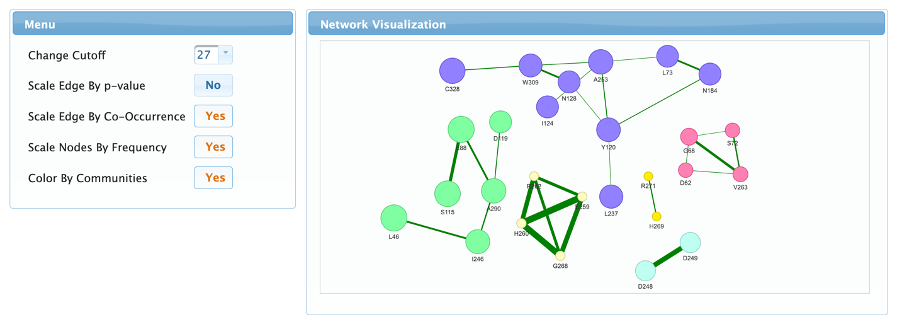
Network page
This page is used to visualize the resulting co-variation network. The network view can be zoomed in and out using the mouse wheel, the entire network or single nodes can be moved by clicking and dragging, and there is also an option to export the image by right clicking and selecting “Save image as”. The left menu allows the user to change five controls on this visualization:
- The network threshold, as in the home page allows the user to change the edges cut-off making the network more stringent or flexible. Changing this value will affect all the results reported (even the ones shown in the other tabs).
- Scale edges by p-value will display the connections thickness according to the negative logarithm of the calculated p-value (probability of the pair of residues occuring randomly in the alignment).
- Scale edges by co-occurrence will display connection thickness according to the Jaccard similarity coefficient calculated from the biadjacency matrix.
- Scale nodes by frequency will display node sizes according to the frequency of the residue in the filtered alignment.
- Color the nodes according to their respective communities.
Conservation page
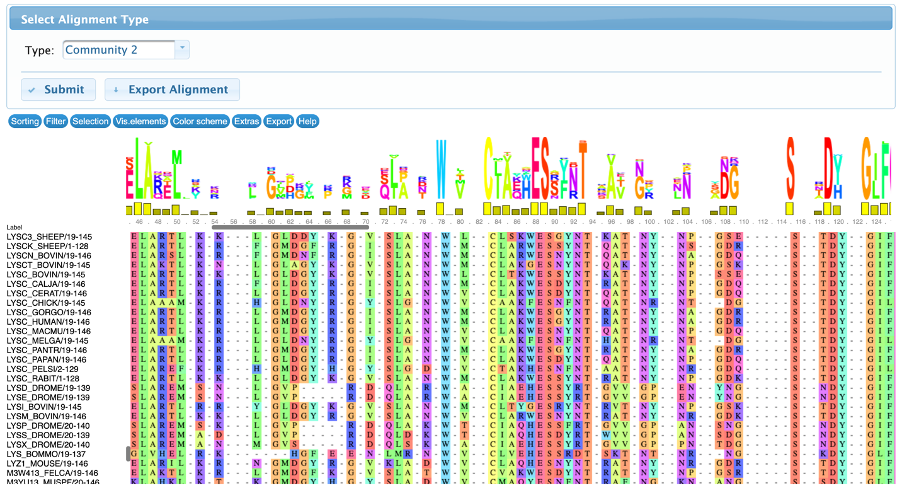
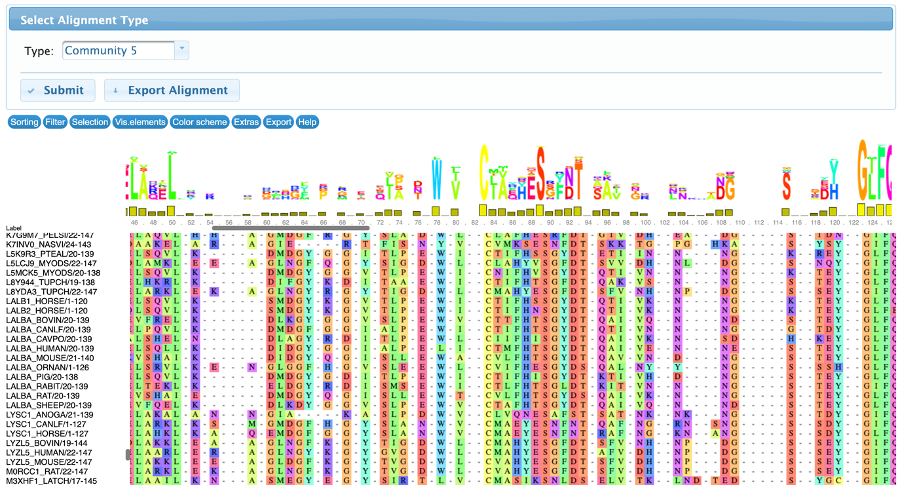
The conservation tab allows the user to visualize and interact with the input multiple sequence alignment and the subalignments of sequences that contains the detected residues. By default, this page displays the resulting alignment after the filtering steps, but it can be changed in the “select alignment type” to show the full alignment (containing all the input sequences, before the filtering steps) or community-based sub-alignments.
There are several controllers divided in seven classes: sorting, filter, selection, visualization, color scheme, extras and export. All these methods are implemented by the MSAViewer package and they do not change the results, therefore are only used for visualization purposes.
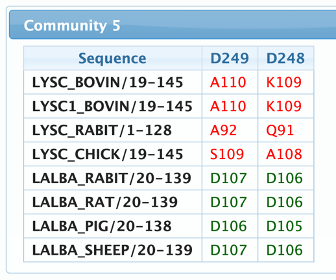
An example can be observed in the figures below. The alignments were sorted by the labels, and it’s possible to see that the residues from the community 2 are present in LYSC sequences and not in LALBA, but the residues of community 5 are present in LALBA sequences and in just a few Lysozymes.
Reference sequences
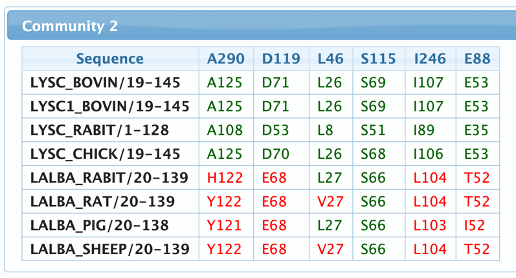
The reference sequences page is used to map and compare how a predefined set of sequences fit in each of the communities. Please be aware that this calculation is performed on the client side, therefore selecting a large number of sequences can require a large amount of RAM.
The table header shows the community residues numbered according to their position in the alignment, then for each selected sequence, the residues are displayed following the numeration of their respective positions in the sequence. Amino acids that match the detected pattern are shown in green, otherwise in red.
Structure page

This page is used to map and visualize the results in a 3D protein structure file. The structure can either be loaded from an input PDB file or directly fetched from the PDBe archive. After submitting the structure, the page will load five visualization reports, as can be seen in the figure below:
- Patterns location – Similarly to the one displayed in the home page, but now showing information about a single sequence, mapped into the structure numbering, and including structural and functional annotations.
- Conservation structure – A dynamic 3D visualization of the submitted structure colored according to the residue conservation in the filtered alignment. The color scale goes from blue (low frequency) to red (high frequency).
- Inter community interactions – This visualization shows predicted structural contacts between residues detected in the analysis. The nodes are colored according to the communities and the edges according to the type of contact.
- Communities structure – Similarly to the conservation structure, but now coloring the residues according to the communities.
- Mapping table – This table maps the positions of the residues in the multiple sequence alignment, protein sequence and structure sequence.
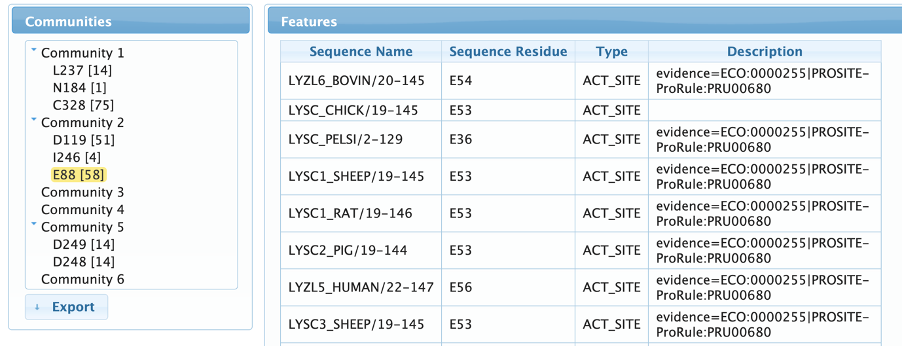
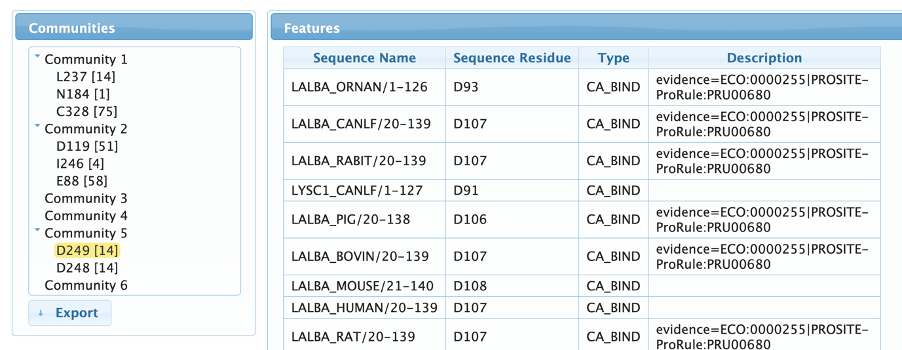
Features page
The features page allows the user to map UniProtKb annotations to the detected residues, guiding the identification of their biological role. The user can navigate through the communities by expanding them in the left menu and clicking on the residues to show all the linked annotation features. As can be observed in the example below, we can clearly see that community 2 contains both active site residues of the C-type Lysozymes, while community 5 contains a pair of aspartates involved in calcium binding, a very important site for alpha-lactoalbumins, but absent in most Lysozymes.
Adherence page
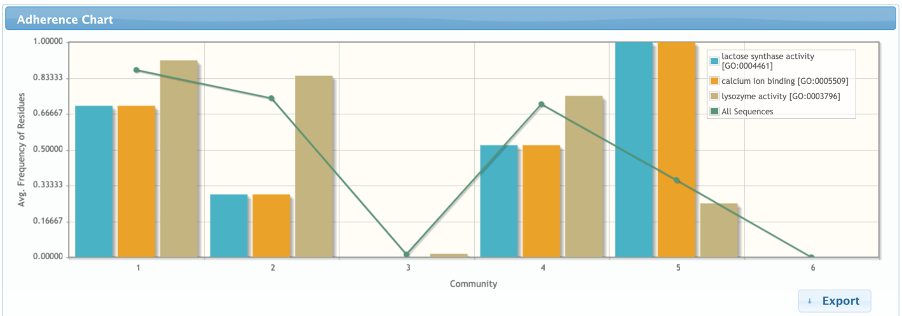
In the adherence page, the user can generate charts comparing how the communities fit to several subsets of sequences defined according to different data types (E.C. number, gene ontologies, INTERPRO term, gene names, protein interaction and involvement in diseases). The user can opt to use only sequences from UniProtKb/Swiss-Prot (manually curated) in this analysis.
The plot shows the average frequency of residues from each community in each subset composed of sequences according to the selected data type. The chart also includes a line that shows the average conservation of the residues of each community in the whole alignment. In the chart below, we can observe that community 2 is a lot more related to the Lysozyme activity, while community 5 is a lot more related to the lactose synthase activity and the calcium binding.
Taxonomy page
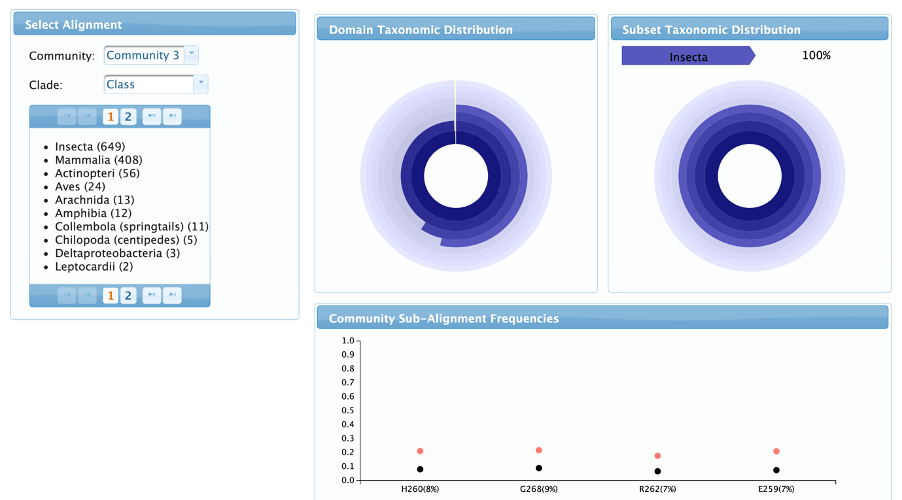
In the taxonomy page it is possible to assess how each community is related taxonomically. The left menu is used to navigate through the communities and taxonomic clades. The user can compare the taxonomic distribution of the subset of sequences that has the residues from the selected community (Subset taxonomic distribution) with the whole alignment taxonomy (Domain taxonomic distribution). The community sub-alignment frequencies chart shows the frequency of each residue in the whole alignment and in the subset of sequenced according to the selected clade.
Example
C-type Lysozyme/Alpha-lactoalbumin
The C-type Lysozyme/Alpha-lactoalbumin protein family, also known as the Glycoside hydrolase family 22 [Davies & Henrissat, 1995], is composed by two well known subfamilies: Alpha-lactoalbumin (LALBA), which regulates lactose production in milk [Qasba, PK et al., 2008] and the C-type Lysozyme (LYSC), an enzyme with bacteriolytic catalytic activity (EC: 3.2.1.17).
Alpha-lactoalbumins lack the catalytic activity of the Lysozymes, but can associate with b-1,4-galactosyl transferase, forming a functional heterodimer called lactose synthase, essential for milk production [Hall & Campbell, 1986]. In addition, all the LALBA's have the ability to bind calcium ions [Stuart et al., 1986], a characteristic that is restricted to only a few LYSC's, such as the equine lysozyme [Nitta, K et al., 1987].
Despite this functional divergence, both C-type Lysozyme and Alpha-lactalbumins are similar in primary and tertiary structure, having probably evolved from a common ancestor Nitta & Sugai, 1989]. Conservation between these groups ranges between 35% and 40%, including the positions of the four disulfide bonds.
This analysis was performed using the Glycoside hydrolase family 22 multiple sequence alignment from Pfam (PF00062). As seen in the seed aligment, this MSA was built using sequences from both LALBAs and Lysozymes, and therefore signals from these two subfamilies are expected to be found in this analysis.
Note that after the alignment is constructed using the hmmerscan program from the HMMER suite it might also contain other closely related homologs and even proteins belonging to uncharacterized groups from the same Protein Family. CONAN default parameters were used in the analysis, including marginal nodes. The results can be accessed here.
The first step usually consists in adjusting the network cut-off by selecting one of the rows in the table at the top of the page. By browsing the community’s co-occurrence matrices, it can be seen that the detected residue pairs display high correlations inside each community. Therefore, the default threshold will be used in this tutorial. This network contains 27 residues grouped in 6 clusters.
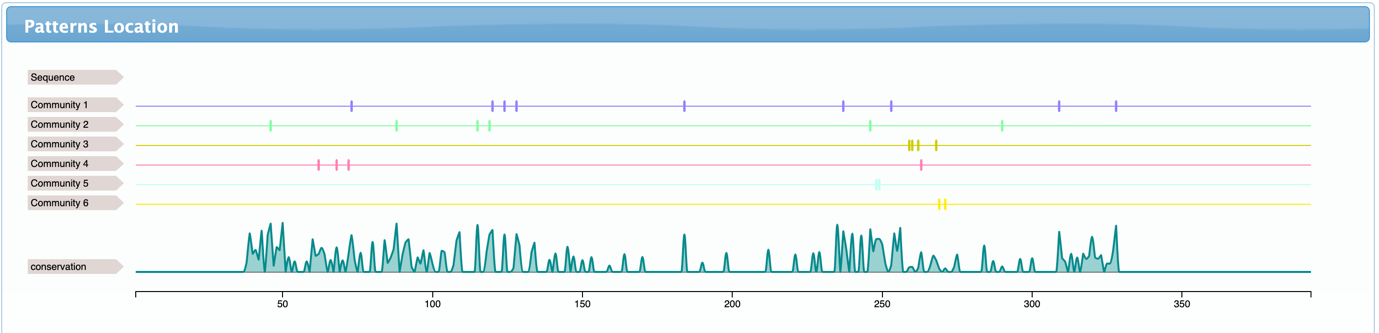
After selecting the network detection threshold, CONAN can be used to guide the identification of the possible biological roles related to the detected coevolving groups. Checking the community-based subalignments is a good start point. If your sequences are labelled in the MSA according to their Uniprot id (gene_species), as in this example, this analysis can map the communities by generating their subalignments and sorting the sequences by their labels. This gives an initial view about the prevalence of the detected coevolving residues. The first insights that we can obtain from visualizing the subalignments are shown below:
- Community 1 and 2 contains several lysozyme sequences and no LALBA
- Community 4 contains several lysozymes sequences and a single rat LALBA
- Community 5 contains LALBA sequences and just a few lysozymes
- Community 3 and 6 do not contain any known lysozyme or LALBA sequence
More solid hypothesis can be raised by checking the correlation between the detected groups and cross-reference annotations present in the manually curated sequences. This analysis can be done in the Features Tab. The chart below was generated by selecting the “Molecular functions” GO and the terms “lactose synthase activity” and “lysozyme activity”.
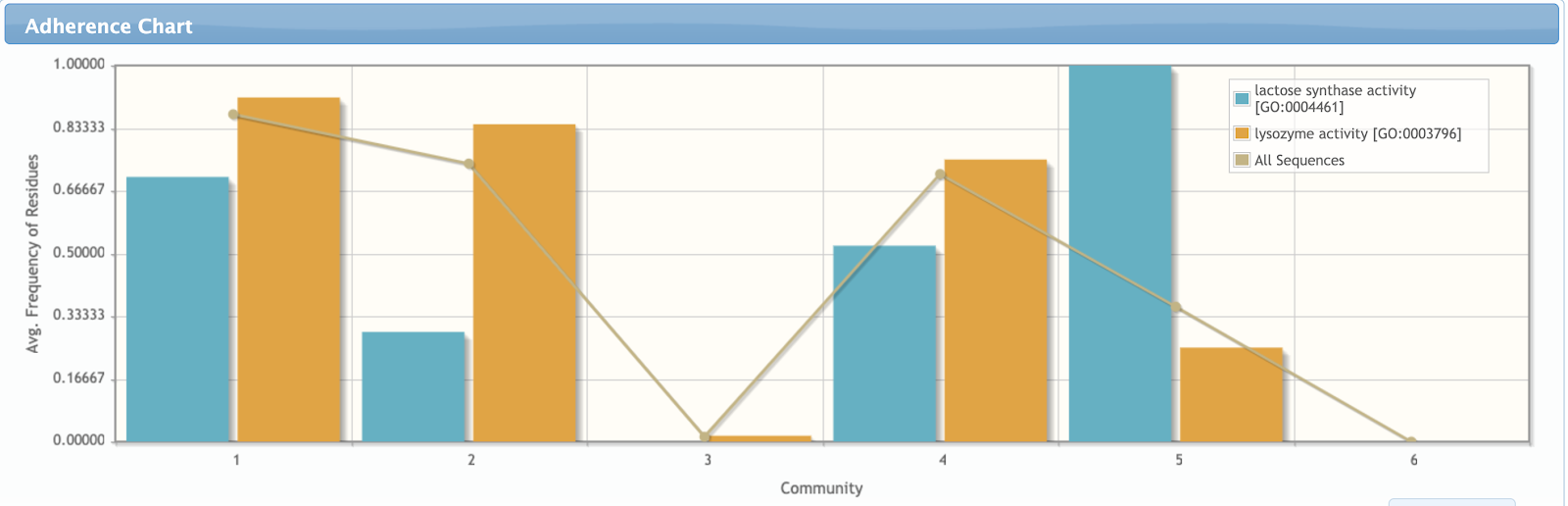
In contrast from what was observed in the subalignments, community 1 does not seem to be specifically related to lysozyme activity. Despite the lack of alpha-lactalbumins in the subset of sequences that contains residues from community 1, it actually includes several Beta-1,4-galactosyltransferases, which are homologues also related to the lactose synthase function. In fact, community 2 is the actual best candidate for determining lysozyme activity, which is paralleled by community 5 for the lactose synthase activity (since this community is conserved in all the lactose synthase proteins from Swiss-prot). Communities 3 and 6 are also interesting, since they are completely unrelated to both lysozyme and lactose synthase activity.
More evidences about the relationship of communities 2 and 5 and enzymatic specificity can be seen in the Features Tab. Community 2 contains the catalytic residues of Lysozymes and community 5 is composed by a pair of aspartic acids involved in calcium binding, a key feature for the alpha-lactalbumins activity.
At this point, the biological roles of communities 3 and 6 are still unknown, but we can get a new insight about them by looking at the Taxonomy Tab: both communities only occur in insect sequences. In fact, there is a group of uncharacterized insect lysozyme-like proteins that lacks the specific catalytic amino acids of the LYSC group and probably have antiviral and antibacterial activities as seen in the literature [Satyavathi, VV et al.,].
These are some basic examples of features that can be easily extracted just by using CONAN, further information can be obtained by a careful and thorough literature review and new experimental information. If you are specific interested in a single sequence of the alignment, you can use the Structure Tab to map the found communities to a structure file (if one is available) and the Reference Sequences Tab to map the detected residues to a set of predefined sequences.
Browser Compatibility
CONAN has been tested on the most widely used web browsers and operating systems. The table below lists the versions of each browser on which this web server was tested.
| 49.0.2623 | 44.0 | n/a | n/a | |
| 71.0.3578 | 64.0 | 80.0.361 | n/a | |
| 80.0.3987 | 73.0.1 | n/a | 13.0.5 | |
| 71.0.3578 | 64.01 | n/a | n/a |




